
Синдром веста что это такое
Синдром Веста – это возрастзависимый эпилептический синдром, относящийся к группе младенческих эпилептических энцефалопатий и характеризующийся особым видом приступов – инфантильными спазмами.
Синдром впервые был описан в 1841 году английским доктором Вестом, когда данное состояние наблюдалось у его ребёнка.
Есть несколько видов синдрома Веста: симптоматический, криптогенный и идиопатический. У каждого вида есть свои характерные ему особенности, течение заболевания и свой прогноз на будущее.
При данном виде заболевания очень важно провести раннюю диагностику, определить диагноз и начать соответствующее лечение, чтобы предотвратить тяжелые когнитивные нарушения. В большинстве случаев, заболевание проявляется у детей в возрасте нескольких месяцев.
Симптомы синдрома Веста
Как правило, синдром проявляется в первые месяцы жизни или в течение первого года жизни ребёнка (до года).
Основным симптомом заболевания являются приступы в виде спазмов. Во время приступа у ребёнка сокращаются различные группы мышц тела и конечностей. Приступы носят серийный характер и, в большинстве случаев, длятся до 15 минут.
Инфантильные спазмы всегда начинаются непредсказуемо и могут возникнуть в любое время суток, но обычно наблюдаются во время пробуждения или засыпания ребёнка. В некоторые случаях, могут наблюдаться генерализованные сокращения мышц, но наиболее частыми являются локальные спазмы.

Причины синдрома Веста
В большинстве случаев, синдром носит симптоматический характер и его причиной является поражение головного мозга по какой-либо причине. Самой распространённой причиной заболевания у детей является гипоксия при родах. Гипоксия – это кислородное голодание головного мозга, которое может возникнуть из-за патологических состояний, при неблагоприятном течении беременности или осложнениях при родах, например, обвитие пуповиной шеи плода.
Помимо этого, к заболеванию могут привести:
- инфекционные заболевания головного мозга в период после рождения;
- структурные патологии головного мозга;
- туберозный склероз;
- генетические и метаболические нарушения;
- структурные изменения в головном мозге;
- ряд других причин.
Внимание родителей! Если причина появления синдрома у вашего ребёнка не обнаружена, крайне важно провести генетические тесты перед планированием следующего ребёнка.
Диагностика
- ЭЭГ – данное исследование крайне важно во время диагностики синдрома Веста, так как на электроэнцефалограмме можно будет заметить определенную, специфическую для заболевания картину.
- МРТ – необходимо для определения наличия поражений головного мозга. В маленьком возрасте данное исследование проводится под наркозом.
- Генетические тесты и метаболические исследования – помогут дать больше информации о вашем случае и подтолкнуть к более эффективному протоколу лечения.
- Дополнительные проверки – они могут быть назначены при необходимости исключения наличия иных заболеваний (о них мы не будет говорить в данной статье, так как это достаточно объемная тема).
ВАЖНО! При первых появлениях настораживающих или подозрительных движений у младенца, следует как можно скорее проконсультироваться с педиатром и неврологом. Обратите внимание, если у малыша наблюдаются регулярные, серийные, цикличные движения. Желательно не ждать очереди даже пару недель!
Иногда родители обращаются за помощью не сразу, особенно если инфантильные спазмы не происходили сериями, что приводит к необративным последствиям.
Лечение синдрома Веста
В большинстве случаев, синдром Веста у детей требуется агрессивное медикаментозное лечение, чтобы предотвратить дальнейшие когнитивные нарушения у ребенка.
Наиболее эффективными противоэпилептическими препаратами при синдроме Веста считаются Сабрил и стероиды в больших дозировках.
Как показывает опыт наших иностранных пациентов, во многих странах мира, в том числе в России, Украине, Казахстане и других странах бывшего СССР, клинические рекомендации могут не включать достаточно высокие дозировки из-за тяжелых побочных эффектов, что, к сожалению, может не дать нужного терапевтического эффекта.
Прогноз
К нам часто поступает вопрос: можно ло ли вылечить синдром Веста и каков прогноз на будущее? Смотрите статистику и факты.
- Примерно у половины детей эпилептические приступы прекратятся.
- В плане моторики у большинства детей не будет проблемы с развитием.
- Симптоматический и криптогенный виды синдрома Веста часто сопровождаются определенной задержкой когнитивного развитии, причем во многих случаях на ранних стадиях тяжело определить тяжесть поражения или его наличие, что затрудняет прогноз.
- У каждого пятого ребёнка с синдромом Веста после наблюдается синдром Леннокса-Гасто.
- В большинстве случаев, дети с данным синдромом страдают от фармакорезистентной эпилепсии, устойчивой к медикаментозной терапии.
Хотите узнать больше о синдроме Веста и необходимом вашему ребёнку лечении?
Подробную информацию о синдроме Веста, диагностике и протоколах лечения можно найти в книге профессора Ури Крамера “Детская эпилепсия от А до Я”. Профессор даёт четкие рекомендации родителям на что нужно обратить внимание на том или ином этапе лечения, а также даёт прогноз развития заболевания в будущем.
Хотите знать, что действительно помогает при эпилепсии и как помочь ребёнку жить максимально полноценной жизнью, несмотря на заболевание?
Раз в неделю мы выпускаем видео или статью о лечении эпилепсии. Это БЕСПЛАТНАЯ и ЕДИНСТВЕННАЯ в своем роде электронная рассылка в мире и мы уверены, что в этих выпусках вы найдёте много полезных рекомендаций для себя и своего ребёнка.
- Можно ли вылечить эпилепсию у детей?
- Переходит ли эпилепсия по наследству?
- Помогают ли альтернативные методы лечения контролировать приступы?
- Что нельзя делать при эпилепсии?
- Сколько должен спать ребёнок с эпилепсией?
- Чем опасны приступы эпилепсии во сне?
- Как помочь пациенту во время приступа?
- Можно ли заниматься спортом?
- Может ли эпилепсия привести к проблемам в учебе, задержке развития, проблем с памятью и поведением?
Введите адрес вашей электронной почты и проверьте почту через 5 минут
***Мы ценим вас и ваше доверие. Наша цель – предоставить вам достоверную информацию о лечении эпилепсии, а также постараться помочь вам или вашим детям жить с этим тяжелым заболеванием. Ни при каких обстоятельствах ваши данные не будут переданы и проданы третьим лицам. Как и вы, мы не любим получать бесполезную почту или рекламу, и постараемся оправдать ваше доверие.

Книга написана простым языком для мам и пап, полна практических советов и рекомендаций эксперта-эпилептолога с мировым именем.
В книге профессора Крамера вы найдёте ответы на многие ваши вопросы об эпилепсии у детей, начиная с видов приступов, правильной диагностики, эффективных методов лечения, и заканчивая практическими советами о том, как повысить качество жизни вашего ребёнка и подготовить его к самостоятельной взрослой жизни.
Синдром Леннокса-Гасто — отдельная форма эпилепсии детского возраста, характеризующаяся наличием полиморфных пароксизмов (миоклонических, атонических, тонических и абсансов) и задержкой нейро-психического развития. Может иметь криптогенный характер или выступать синдромом других патологических состояний (церебральных аномалий, генетических обменных заболеваний, перинатальной патологии). Синдром Леннокса-Гасто диагностируется по типичной вариативной картине эпиприступов и характерному паттерну электроэнцефалограммы. Дополнительно проводится МРТ и КТ головного мозга. Антиконвульсантная терапия синдрома малоэффективна, проводится поиск альтернативных методов лечения. Прогноз вариабельный, но в большинстве случаев неблагоприятный.
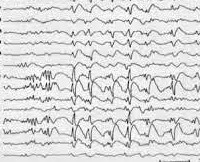
Общие сведения
Синдром Леннокса-Гасто (СЛГ) — вариант эпилепсии детского возраста, для которого характерно сочетание атонических, миоклонических, тонических эпиприступов и атипичных абсансов, медленный островолновой паттерн ЭЭГ. В 1950 г. СЛГ был выделен в качестве отдельного эпилептического синдрома, а в 1964-1966 гг. неврологическое сообщество признала его самостоятельной нозологической формой. Синдром Леннокса-Гасто по различным данным составляет от 3% до 10% всех случаев детской эпилепсии. Его распространенность колеблется в пределах 1-2,8 случаев на 10 тыс. Несколько чаще встречается у мальчиков. Типичный возраст начала заболевания от 2 до 5 лет, реже — 6-8 лет. Сегодня СЛГ является тяжелым заболеванием с прогрессирующим течением, эффективное лечение которого пока является предметом надежд многих специалистов в области детской неврологии и эпилептологии.
Причины синдрома Леннокса-Гасто
Синдром Леннокса-Гасто относится к заболеваниям, этиологические факторы которых пока точно не установлены. Известно, что во многих случаях синдром носит симптоматический характер и формируется на фоне генетической патологии, последствий различных неблагоприятных факторов, действующих в перинатальном периоде и на 1-ом году жизни. Однако в большинстве случаев морфологический субстрат заболевания остается не выявленным. К этиофакторам, способным спровоцировать развитие СЛГ, относят гипоксию плода, внутриутробные инфекции (краснуху, цитомегалию, герпес, токсоплазмоз), родовые травмы новорожденных (в первую очередь внутричерепные), недоношенность, асфиксию новорожденных, тяжелые инфекционные заболевания постнатального периода (менингит, энцефалит), аномалии развития головного мозга (гидроцефалию, кортикальную дисплазию, гипоплазию мозолистого тела и др.), метаболические нарушения с поражением ЦНС, отдельные генетические заболевания (например, туберозный склероз).
В 25-40% случаев синдром Леннокса-Гасто возникает у детей с отягощенным по эпилепсии семейным анамнезом. Кроме того, существует гипотеза об этиологической роли иммунных нарушений, в т. ч. возникающих вследствие вакцинации. Примерно в 30% случаев СЛГ является следствием эволюции синдрома Веста. Когда синдром Леннокса-Гасто манифестирует на фоне полного благополучия в здоровье ребенка и отсутствия в его анамнезе вышеперечисленных факторов, говорят о криптогенной (не имеющей вероятной причины) форме заболевания. Криптогенный вариант СЛГ встречается в 10-20% случаев и отличается более благоприятным течением.
Симптомы синдром Леннокса-Гасто
Симптоматический синдром Леннокса-Гасто, как правило, дебютирует на фоне уже имеющегося отставания в умственном и психическом развитии. При криптогенной форме развитие ребенка на момент манифестации синдрома соответствует норме. СЛГ отличается большой вариативностью приступов, их различной продолжительностью и частотой.
Миоклонические пароксизмы представляют собой локальные мышечные подергивания. Чаще охватывают мышцы-сгибатели проксимальных отделов рук, при распространении на нижние конечности происходит падение. Характеризуются симметричным серийным возникновением в обеих конечностях и стереотипностью. Нуждаются в дифференцировке с миоклониями при клещевом энцефалите и токсических поражениях ЦНС; миоклонусом неэпилептического характера, для которого типичны нерегулярные асимметричные миоклонии, возникающие в ответ на различные сенсорные раздражители (звук, свет, прикосновение) и не сопровождающиеся изменениями ЭЭГ.
Тонические пароксизмы СЛГ часто возникают в период сна и отличаются своей кратковременностью (средняя длительность 10 сек.). Сопровождаются отключением сознания. Могут иметь генерализованный характер или проявляться в виде тонического напряжения отдельных мышечных групп (заднешейных, спинных, мышц брюшного пресса, плечевого пояса и пр.). Тонические пароксизмы сопровождаются тахикардией, цианозом лица, слезотечением, апноэ, гиперсаливацией. Минимальные локальные пароксизмы тонического характера иногда с трудом можно отдифференцировать от зевоты или потягивания.
Задержка психомоторного развития (ЗПР) отмечается почти во всех случаях СЛГ. Ее выраженность зависит от формы синдрома (криптогенная или симптоматическая), характера фоновой патологии ЦНС, тяжести и частоты эпилептических пароксизмов. Как правило, на первый план выходят проблемы с обучением владению новыми навыками и с усвоением новой информации. Зачастую наблюдается агрессивность, гиперактивность, эмоциональная нестабильность, характерные для аутизма особенности характера. Около 50% подростков, имеющих синдром Леннокса-Гасто, не владеют навыками самообслуживания. Еще 25% социально и эмоционально дезадаптированы по причине выраженной олигофрении. Особенности поведения и характера не дают возможность нормально адаптироваться в социуме даже тем пациентам, у которых олигофрения имеет легкую степень выраженности. Нормальная социальная адаптация наблюдается лишь в 15% случаев.
Диагностика синдрома Леннокса-Гасто
Синдром Леннокса-Гасто устанавливается на основании типичной клинической картины, состоящей из полиморфных эпиприступов и симптомов отставания нейро-психического развития. Учитывается также возраст начала пароксизмов и семейный эпилептический анамнез. Большую диагностическую роль играет электроэнцефалография. Межприступная (интериктальная) ЭЭГ в бодрствующем состоянии регистрирует плохую структурированность и замедленность основного ритма. ЭЭГ-паттерн имеет картину гипсаритмии с большим количеством спайков различной амплитуды. Наиболее высокие пики регистрируются в лобной области. ЭЭГ-паттерн в период приступов зависит от их формы.
Методы нейровизуализации (МРТ и КТ головного мозга) выявляют преимущественно неспецифичные патологические изменения: внутреннюю гидроцефалию, атрофию подкорковых областей и корковых структур преимущественно лобной зоны, гипоплазию лобных долей. Попытки проанализировать при помощи ПЭТ головного мозга степень утилизации глюкозы церебральными тканями дали противоречивые сведенья: в одних случаях были выявлены зоны гиперметаболизма, в других — гипометаболизма; у части пациентов метаболизм глюкозы был в пределах нормы.
По причине большой вариативности пароксизмов, синдром Леннокса-Гасто следует дифференцировать с целым рядом других форм эпилепсии, дебютирующих в детском возрасте: с миоклонической эпилепсией, доброкачественной роландической эпилепсией, синдромом Веста, детской абсансной эпилепсией, дисметаболической эпилепсией при болезни Гоше, Краббе, Ниманна-Пика и др.
Лечение синдрома Леннокса-Гасто
Терапия проводится противоэпилептическими средствами. Применяются вальпроевая к-та, этосуксимид, карбамазепин, ламотриджин и др. В большинстве случаев проводится комбинированное лечение одним из указанных фармпрепаратов и вальпроатом натрия. Однако до 90% случаев синдрома Леннокса-Гасто являются резистентными к антиконвульсантной терапии. В связи с этим основной целью лечения является уменьшение числа эпиприступов и улучшение качества жизни ребенка и его семьи в межпароксизмальный период.
Неврологами и эпилептологами ведется поиск новых способов терапии. Доказанной является положительная роль кетогенной диеты, заключающейся в резком ограничении употребления углеводов и повышении содержания жиров в пище. Рядом клиницистов отмечен положительный эффект лечения синдрома Леннокса-Гасто большими дозировками иммуноглобулина. Наблюдалась эффективность применения АКТГ и глюкокортикоидов. В случаях, когда синдром Леннокса-Гасто сопровождается частыми и тяжелыми эпипароксизмами с падением и угрозой травматизации ребенка, совместно с нейрохирургом может быть рассмотрен вопрос о проведении хирургической операции рассечения мозолистого тела — каллозотомии. Подобное вмешательство не избавляет пациентов от приступов, но существенно уменьшает их интенсивность.
К новым способам лечения относится имплантация стимулятора блуждающего нерва и RNS-стимулятора. В первом случае прибор устанавливается подкожно в область ключицы, а его электрод проводят к проходящему в шее блуждающему нерву. По данным проведенных в США и Европе исследований, в 60% случаев данное устройство позволяет снизить количество эпиприступов. Во втором случае прибор вшивается под кожу головы, а его электроды имплантируются в зону эпилептогенного очага. С их помощью, подобно ЭЭГ, устройство постоянно регистрирует электрическую активность мозга. При получении сигналов, свидетельствующих о начинающемся пароксизме, прибор генерирует ответные импульсы, обеспечивающие супрессию эпилептической активности.
Прогноз синдрома Леннокса-Гасто
Синдром Леннокса-Гасто имеет в основном неблагоприятный прогноз. До 10% случаев заканчивается гибелью детей в течение первого десятилетия жизни. Летальные исходы связаны преимущественно с тяжелой травматизацией во время эпиприступов с падением. Прогностически неблагоприятными критериями считаются: манифестация синдрома в более раннем возрасте, начало судорог на фоне ЗПР, предшествующий синдром Веста, высокая частота и интенсивность пароксизмов. Невозможность медикаментозного купирования эпиприступов приводит к прогрессирующей ЗПР. Практически у всех пациентов наблюдается выраженная в различной степени умственная отсталость, половина больных не способны к самообслуживанию.
Синдром Веста — серийные спастические сокращения в отдельных мышечных группах или генерализованного характера, протекающие на фоне задержки нейропсихического развития и сопровождающиеся гипсаритмическим ЭЭГ-паттерном. Манифестирует в возрасте до 4-х лет, преимущественно на 1-ом году жизни. В большинстве случаев имеет симптоматический характер. Диагностика синдрома основана на клинических данных и результатах ЭЭГ. Для выявления основной патологии необходимы КТ или МРТ, ПЭТ головного мозга, консультация генетика, нейрохирурга. Лечение возможно противоэпилептическими препаратами, стероидами (АКТГ, преднизолон), вигабатрином. По показаниям решается вопрос о хирургическом лечении (каллозотомия, удаление патологического очага).
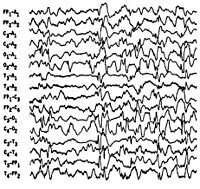
Общие сведения
Синдром Веста носит название по имени врача, наблюдавшего его проявления у своего ребенка и впервые описавшего его в 1841 г. В связи с манифестацией синдрома в раннем возрасте и протеканием судорог по типу серии отдельных спазмов, пароксизмы, характеризующие синдром Веста, получили название инфантильные спазмы. Первоначально заболевание относили к генерализованной эпилепсии. В 1952 г. был изучен специфический гипсаритмический ЭЭГ-паттерн, патогномоничный для этой формы эпилепсии и характеризующийся медленноволновой асинхронной активностью с беспорядочными спайками высокой амплитуды. В 1964 г. специалистами в области неврологии синдром Веста был выделен в качестве отдельной нозологии.
Внедрение в неврологическую практику нейровизуализации позволило определить наличие у пациентов очаговых поражений вещества мозга. Это заставило неврологов пересмотреть свои взгляды на синдром Веста как на генерализованную эпилепсию и отнести его в ряд эпилептических энцефалопатий. В 1984 г. был выявлена эволюция эпилептической формы энцефалопатии от ее раннего варианта в синдром Веста, а с течением времени в синдром Леннокса-Гасто.
В настоящее время синдром Веста занимает около 2% от всех случаев эпилепсии у детей и примерно четверть младенческой эпилепсии. Распространенность составляет, по различным источникам, от 2 до 4,5 случаев на 10 тыс. новорожденных. Несколько чаще заболевают мальчики (60%). 90% случаев манифестации синдрома приходится на 1-й год жизни, с пиком в возрасте от 4 до 6 мес. Как правило, к возрасту 3-х лет мышечные спазмы проходят или трансформируются в иные формы эпилепсии.
Причины синдрома Веста
В подавляющем большинстве случаев синдром Веста носит симптоматический характер. Он может возникать вследствие перенесенных внутриутробных инфекций (цитомегалии, герпетической инфекции), постнатального энцефалита, гипоксии плода, преждевременных родов, внутричерепной родовой травмы, асфиксии новорожденного, постнатальной ишемии вследствие позднего пережатия пуповины. Синдром Веста может являться следствием аномалий строения головного мозга: септальной дисплазии, гемимегалоэнцефалии, агенезии мозолистого тела и пр. В ряде случаев инфантильные спазмы выступают симптомом факоматозов (синдрома недержания пигмента, туберозного склероза, нейрофиброматоза), точечных генных мутаций или хромосомных аберраций (в т. ч. синдрома Дауна). В литературе упоминаются случаи фенилкетонурии с инфантильными спазмами.
В 9-15% синдром Веста является идиопатическим или криптогенным, т. е. его первопричина не установлена или не очевидна. Зачастую при этом прослеживается наличие случаев фибрильных судорог или эпиприступов в семейном анамнезе больного ребенка, т. е. имеет место наследственная предрасположенность. Ряд исследователей указывают, что фактором, провоцирующим синдром Веста, может выступать вакцинация, в частности введение АКДС. Это может быть связано с совпадением сроков вакцинации и возраста типичного дебюта синдрома. Однако достоверные данные, подтверждающие провоцирующую роль вакцин, до сих пор получены.
Симптомы синдрома Веста
Как правило, симптом Веста дебютирует на первом году жизни. В отдельных случаях его манифестация происходит в более старшем возрасте, однако не позже 4-х лет. Основу клиники составляют серийные мышечные спазмы и нарушение психомоторного развития. Первые пароксизмы зачастую появляются на фоне уже существующей задержки психомоторного развития (ЗПР), но в 1/3 случаев возникают у первично здоровых детей. Отклонения в нейропсихологическом развитии наиболее часто проявляются снижением и выпадением хватательного рефлекса, аксональной гипотонией. Возможно отсутствие слежения глазами за предметами и расстройство фиксации взора, что является прогностически неблагоприятным критерием.
Мышечные спазмы носят внезапный симметричный и кратковременный характер. Типична их серийность, при этом интервал между следующими друг за другом спазмами длится не менее 1 минуты. Обычно наблюдается возрастание интенсивности спазмов в начале пароксизма и ее спад в конце. Число спазмов, происходящих за сутки, варьирует от единиц до сотен. Наиболее часто возникновение инфантильных спазмов происходит в период засыпания или сразу после сна. Провоцировать пароксизм способны резкие громкие звуки и тактильная стимуляция.
Семиотика пароксизмов, которыми сопровождается синдром Веста, зависит от того, какая мышечная группа сокращается — экстензорная (разгибательная) или флексорная (сгибательная). По этому признаку спазмы классифицируют на экстензорные, флексорные и смешанные. Чаще всего наблюдаются смешанные спазмы, затем сгибательные, наиболее редко — разгибательные. В большинстве случаев у одного ребенка наблюдаются спазмы нескольких видов и то, какой именно спазм будет преобладать, зависит от положения тела в момент начала пароксизма.
Наряду с серийными спазмами, синдром Веста может сопровождаться бессудорожными приступами, проявляющимися внезапной остановкой двигательной активности. Иногда отмечаются пароксизмы, ограниченные подергиванием глазных яблок. Возможно нарушение дыхания вследствие спазма дыхательной мускулатуры. В некоторых случаях имеют место асимметричные спазмы, проявляющиеся отведением головы и глаз в сторону. Могут встречаться и другие виды эпиприступов: фокальные и клонические. Они комбинируются со спазмами или имеют самостоятельный характер.
Диагностика синдрома Веста
Синдром Веста диагностируется по основной триаде признаков: приступы кластерных мышечных спазмов, задержка психомоторного развития и гипсаритмический ЭЭГ-паттерн. Имеют значение возраст манифестации спазмов и их связь со сном. Трудности диагностики возникают при позднем дебюте синдрома. В ходе диагностики ребенок консультируется педиатром, детским неврологом, эпилептологом, генетиком. Дифференцировать синдром Веста следует с доброкачественным младенческим миоклонусом, доброкачественной роландической эпилепсией, младенческой миоклонической эпилепсией, синдромом Сандифера (наклон головы по типу кривошеи, гастроэзофагальный рефлюкс, эпизоды опистотонуса, которые могут быть приняты за спазмы).
КТ головного мозга у имеющих синдром Веста детей может выявлять диффузные либо очаговые изменения церебральных структур, но может быть в пределах нормы. В диагностике локальных поражений более чувствительным методом является МРТ головного мозга. Для выявления участков гипометаболизма мозговых тканей в некоторых случаях возможно проведение ПЭТ головного мозга.
Лечение синдрома Веста
Синдром Веста считался резистентным к проводимой терапии вплоть до открытия в 1958 г. влияния на приступы препаратов АКТГ. Терапия АКТГ и преднизолоном приводит к значительному улучшению или полному прекращению инфантильных спазмов, что сопровождается исчезновением гипсаритмического ЭЭГ-паттерна. До сих пор среди неврологов нет однозначных решений касательно доз и длительности стероидной терапии. Исследования показали, что в 90% случаев терапевтический успех достигался при применении больших дозировок АКТГ. Сроки терапии могут варьировать в пределах 2-6 недель.
Новый этап в лечении инфантильных спазмов начался в 1990-1992 гг. после обнаружения положительного терапевтического эффекта вигабатрина. Однако преимущество лечения вигабатрином пока доказано лишь для больных туберозным склерозом. В остальных случаях исследования показали большую эффективность стероидов. С другой стороны стероидная терапия имеет худшую, в сравнении с вигабатрином, переносимость и более высокий процент рецидивов.
Из антиконвульсантов эффективность показана лишь у нитразепама и вальпроевой кислоты. У отдельных пациентов описан лечебный эффект больших доз витамина В6, который отмечался в первые недели терапии. При инфантильных спазмах, резистентных к проводимой терапии, с подтвержденным на томографии наличием патологического очага показана консультация нейрохирурга для решения вопроса о резекции очага. Если подобная операция невозможна, то при наличии дроп-атак проводится тотальная каллозотомия (пересечение мозолистого тела).
Прогноз синдрома Веста
Обычно к 3-летнему возрасту наблюдается регресс и исчезновение инфантильных спазмов. Но примерно в 55-60% случаев они трансформируются в другую форму эпилепсии, чаще всего в синдром Леннокса-Гасто. Фармакорезистентность часто констатируется при инфантильных спазмах, сопровождающих синдром Дауна. Даже при успешном купировании пароксизмов синдром Веста имеет неудовлетворительный прогноз в плане психомоторного развития ребенка. Возможны когнитивные и поведенческие нарушения, ДЦП, аутизм, трудности в обучении. Остаточный психомоторный дефицит не наблюдается только в 5-12% случаев. ЗПР отмечается у 70-78% детей, двигательные расстройства — у 50%. Серьезный прогноз имеет синдром Веста, обусловленный аномалиями или дегенеративными изменениями головного мозга. При этом летальность может достигать 25%.
Более благоприятный прогноз имеют криптогенный и идиопатический синдром Веста при отсутствии ЗПР до появления спазмов. В этой группе больных остаточный интеллектуальный или неврологический дефицит отсутствует у 37-44% детей. Неблагоприятно отражается на прогнозе болезни откладывание начала лечения. Прогностическая оценка затрудняется тем, что отдаленные последствия также зависят от основной патологии, на фоне которой возникает симптоматический синдром Веста.
Вестибулярная атаксия — нарушения координации движений и способности поддерживать позу, связанные с поражением вестибулярного аппарата на любом его уровне. Вестибулярная атаксия проявляется шаткостью в положении стоя и сидя, а также при ходьбе. Она сопровождается системным головокружением и нистагмом; могут наблюдаться тошнота и рвота, вегетативные нарушения и симптомы, характерные для того патологического процесса, который послужил причиной развития вестибулярной атаксии. Диагностика последнего является главной целью обследования пациентов с вестибулярной атаксией. Лечение вестибулярной атаксии является симптоматическим. Основная терапия должна быть направлена на причинное заболевание.
МКБ-10

Общие сведения
Ориентацию тела в пространстве в организме человека обеспечивает вестибулярный анализатор. Он отвечает за определение положения и характера движения тела и отдельных его частей, обеспечивает восприятие силы тяжести. Любое изменение положения тела в пространстве воспринимается вестибулярными рецепторами — так называемыми волосковыми клетками, расположенными в лабиринте внутреннего уха. От рецепторов нервные импульсы идут по вестибулярному нерву, который вместе со слуховым нервом входит в VIII пару черепно-мозговых нервов. Далее импульсы поступают в вестибулярные ядра продолговатого мозга, где происходит синтез информации и осуществляется управление двигательными реакциями. Из вестибулярных ядер регулирующие нервные импульсы расходятся в различные отделы ЦНС: мозжечок, спинной мозг, ретикулярную формацию, вегетативные нервные узлы, глазодвигательные ядра и кору головного мозга. Они обеспечивают перераспределение мышечного тонуса и рефлекторные реакции по сохранению равновесия.
Причины
Вестибулярная атаксия связана с поражением любой структуры вестибулярного анализатора. Чаще всего она вызвана повреждением волосковых клеток в результате воспалительного процесса во внутреннем ухе — лабиринтита. В свою очередь лабиринтит может возникать в результате травмы уха или при переходе инфекции из полости среднего уха при остром среднем отите, хроническом гнойном среднем отите, осложненном аэроотите. Гибель волосковых клеток, приводящая к развитию вестибулярной атаксии, может произойти в результате инвазивного роста опухоли уха или токсического воздействия выделений холестеатомы уха. Приступообразная вестибулярная атаксия сопровождает болезнь Меньера.
Реже вестибулярная атаксия бывает вызвана поражением вестибулярного нерва, которое может иметь инфекционный, опухолевый (при невриноме слухового нерва) или токсический (при приеме ототоксических лекарственных средств) характер. Зачастую вестибулярный нейронит связан с вирусной инфекцией: ОРВИ, вирусом герпеса, гриппом и др.
Вестибулярная атаксия может возникнуть при поражении расположенных в продолговатом мозге вестибулярных ядер. Так, вестибулярная атаксия возникает в результате сдавления продолговатого мозга у лиц с краниовертебральными аномалиями (аномалией Киари, ассимиляцией атланта, платибазией), при опухолях ствола мозга, энцефалите и арахноидите задней черепной ямки, демиелинизирующих заболеваниях (рассеянном склерозе, остром энцефаломиелите). Вестибулярная атаксия является клиническим проявлением хронической ишемии ствола мозга, обусловленной нарушением вертебро-базилярного кровообращения в связи с синдромом позвоночной артерии, атеросклерозом, гипертонической болезнью, аневризмой сосудов головного мозга. При острых нарушениях кровообращения этой области (ТИА, ишемическом или геморрагическом инсульте) также наблюдается вестибулярная атаксия.
Вестибулярная атаксия зачастую наблюдается после перенесенной черепно-мозговой травмы. При этом она может быть вызвана как непосредственным воздействием травмирующего фактора на ядра и корешки вестибулярного нерва, так и с сопутствующими травме нарушениями кровообращения (посттравматический сосудистый спазм).
Симптомы вестибулярной атаксии
Вестибулярная атаксия проявляется как в движении (динамическая атаксия), так и в положении стоя (статическая атаксия). От других видов атаксии вестибулярная атаксия отличается зависимостью ее выраженности от поворотов головы и туловища. Усиление атаксии при поворотах головы, глаз и туловища заставляют пациентов избегать подобных движений или выполнять их плавно и медленно. Зрительный контроль движений частично компенсирует нарушения функции вестибулярного анализатора, поэтому с закрытыми глазами пациент чувствует себя более неуверенно и проявления вестибулярной атаксии нарастают.
Поражения вестибулярного анализатора чаще всего носят односторонний характер. В таких случаях вестибулярная атаксия проявляется шаткостью при ходьбе с отклонением тела постоянно в одну и ту же сторону — в сторону, где локализуется очаг поражения. В позе стоя или сидя пациент также отклоняется в пораженную сторону. Этот симптом легко выявляется в позе Ромберга и при попытке пациента пройти несколько шагов ровно с закрытыми глазами.
Характерным признаком вестибулярной атаксии является наличие системного головокружения, при котором пациент испытывает чувство вращения собственного тела или движения вокруг себя окружающих предметов. Головокружение может отмечаться даже в положении лежа с закрытыми глазами. В таких случаях оно обычно сопровождается нарушением сна с затруднениями при засыпании. Срабатывание вестибуло-висцеральных нервных связей приводит к тому, что головокружение при вестибулярной атаксии зачастую сопровождается тошнотой и рвотой. Вестибуло-вегетативные взаимодействия обуславливают появление вегетативных реакций: бледности или красноты лица, чувства страха, тахикардии, лабильности пульса, гипергидроза.
Во большинстве случаев вестибулярная атаксия сопровождается горизонтальным нистагмом, направление которого противоположно стороне поражения. Возможен билатеральный нистагм. При поражении вестибулярных ядер может отмечаться вертикальный нистагм с ротаторным компонентом. При нарушении на уровне периферического отдела вестибулярного анализатора нистагм усиливается при поворотах головы, однако при повторных поворотах нистагм может уменьшаться. При наличии краниовертебральной аномалии вестибулярная атаксия сопровождается нистагмом, который усиливается при наклонах головы.
Диагностика
Вестибулярная атаксия может быть выявлена по характерным жалобам пациента и в ходе его неврологического осмотра. Для дифференцировки вестибулярной атаксии от атаксий других видов (мозжечковой, сенсетивной, корковой), а также для установления уровня и характера поражения вестибулярного анализатора, неврологу необходимы результаты инструментальных методов обследования: РЭГ, Эхо-ЭГ, ЭЭГ, КТ и МРТ головного мозга, рентгенологические исследования. Поскольку вестибулярная атаксия как синдром встречается при многих заболеваниях ЦНС, важнейшим моментом в клинической неврологии является выявление причины ее развития.
РЭГ позволяет получить косвенные данные о состоянии кровообращения головного мозга. При необходимости она может быть дополнена ангиографией или МРТ ангиографией сосудов головного мозга. С помощью Эхо-ЭГ оценивают состояние ликворной системы головного мозга. Смещение Эхо свидетельствует о наличие объемного образования (опухоли, гематомы или абсцесса головного мозга), с наличием которого может быть связана вестибулярная атаксия. ЭЭГ используют для анализа биоэлектрической активности мозга. КТ и МРТ позволяют диагностировать объемные образования и демиелинизирующие процессы, которые также могут быть причиной вестибулярной атаксии. При подозрении на краниовертебральную аномалию проводят рентгенографию позвоночника в шейном отделе и рентгенографию черепа.
Исследование вестибулярного анализатора у пациентов с вестибулярной атаксией проводит вестибулолог, а при его отсутствии невролог или отоларинголог. Обследование включает проведение вестибулометрии, стабилографии, видеоокулографии или электронистагмографии, а также калорической пробы. Поскольку вестибулярная атаксия зачастую сочетается с нарушениями слуха (тугоухостью), то проводится исследование слуха: пороговая аудиометрия, исследование камертоном, электрокохлеография, промонториальный тест и др.
Лечение вестибулярной атаксии
Терапия вестибулярной атаксии направлена в первую очередь на излечение вызвавшего ее заболевания. При наличии инфекционного поражения уха проводят антибиотикотерапию, промывание среднего уха, санирующую операцию, лабиринтотомию и пр. мероприятия, направленные на ликвидацию гнойного очага. Если вестибулярная атаксия обусловлена сосудистыми нарушениями, то применяют препараты, улучшающие кровоснабжение головного мозга. В тяжелых случаях краниовертебральных аномалий проводится их хирургическая коррекция. Соответствующая терапия необходима при наличии объемных образований, энцефалита, арахноидита.
Сама вестибулярная атаксия подлежит симптоматическому лечению, как правило заключающемуся в приеме препаратов, улучшающих метаболизм и функционирование нервных клеток: пирацетама, гамма-аминомасляной кислоты, гинкго билоба, витаминов группы В. Кроме того, пациентам рекомендован специальный комплекс ЛФК, направленный на тренировку координации движений и укрепление мышц.
Эпилептическая энцефалопатия – это вид эпилепсии, проявления которой могут привести к повреждению головного мозга.
Эпилептическая энцефалопатия является тяжелым вариантом течения заболевания, при котором, в большинстве случаев, наблюдаются не только эпилептические приступы, но и когнитивные нарушения различной степени тяжести.
Дебютируют данные заболевания, в большинстве случаев, в раннем возрасте на фоне нормального или уже имеющегося нарушенного развития. Медицине известно несколько десятков различных ЭЭ, с наиболее часто встречающимися из них мы познакомим вас в данной статье.
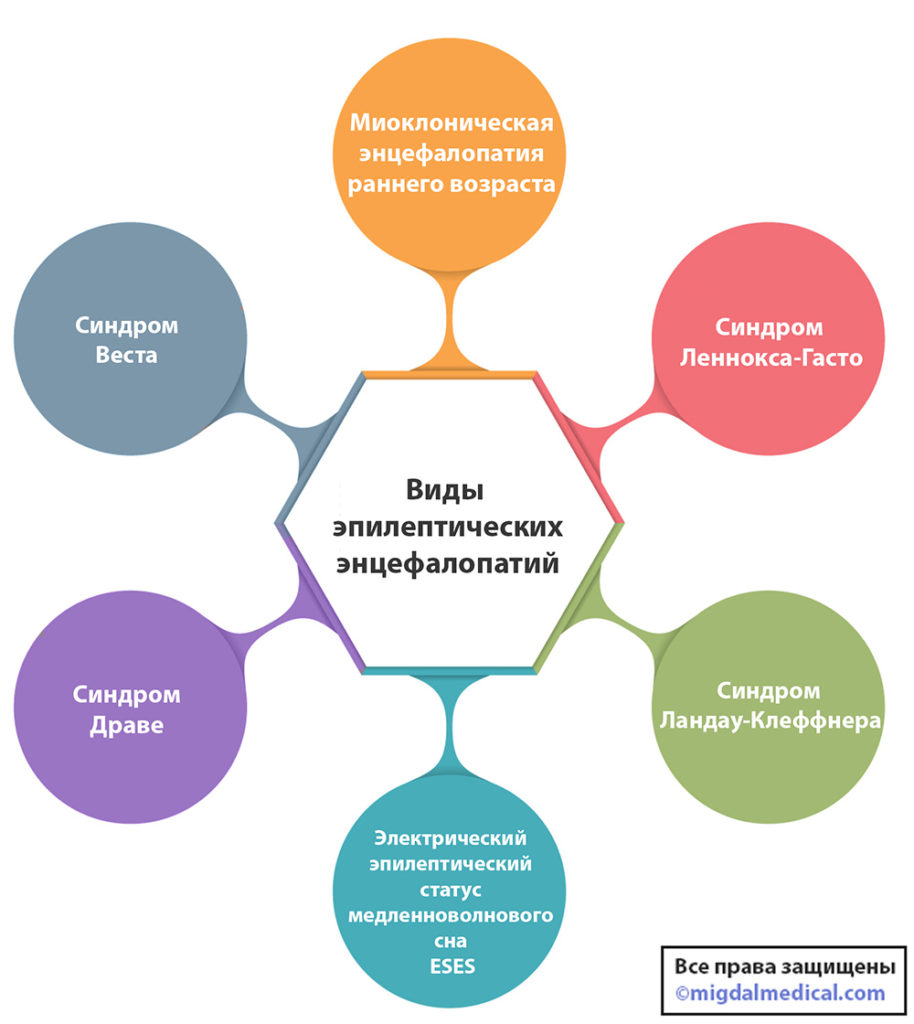
Виды эпилептических энцефалопатий
- Миоклоническая энцефалопатия раннего возраста
Ранняя миоклоническая энцефалопатия является редкой возраст-зависимой формой эпилепсии. Диагноз может быть установлен посредством ЭЭГ, так как на нём проявляется специфическая эпилептическая активность головного мозга, свойственная именно данной ЭЭ. К сожалению, данный тип болезни, в большинстве случаев, является устойчивым к медикаментозному лечению противоэпилептическими препаратами.
Синдром Веста – это возрастзависимый эпилептический синдром, относящийся к группе младенческих эпилептических энцефалопатий и характеризующийся особым видом приступов – инфантильными спазмами. Данное заболевание может привести к серьезным когнитивным нарушениям, а потому крайне важно как можно раньше провести диагностику и назначить подходящее пациенту лечение.
Причиной синдрома Веста является поражение головного мозга, которое наиболее часто появляется во время гипоксии (кислородного голодания головного мозга) при родах. Также причиной возникновения повреждений могут стать инфекционные заболевания, структурные патологии мозга, генетические нарушения и другие.
В большинстве случаев, синдром Веста у детей требуется агрессивное медикаментозное лечение, чтобы предотвратить дальнейшие когнитивные нарушения у ребенка. Узнать больше о синдроме и его лечении вы можете на следующей странице – Синдром Веста.
Синдром Драве – это детская эпилептическая энцефалопатия наследственного характера, которая проявляется эпилептиформными приступами и приводит к когнитивным нарушениям. От данного заболевания чаще страдают девочки, нежели мальчики. Как правило, до 5 месяцев родители не замечают каких-либо отклонений в развитии ребёнка и у него ответствуют судороги, однако после 5 месяцев энцефалопатия даёт о себе знать.
Во время синдрома у ребёнка наблюдается большое количество разнообразных приступов: фокальные, фебрильные, миоклонические и генерализованные судорожные припадки. Помимо этого, у детей проявляются другие синдромы: проблемы с моторикой, тремор, дизартрия, атаксия и другие.
К сожалению, большинство детей, страдающих данным синдромом, не восприимчивы к медикаментозной терапии. Однако, в некоторых случаях удаётся достичь снижения количества приступов благодаря противоэпилептическим препаратам.
Больше узнать о данном синдроме можно на следующей странице – Синдром Драве.
Синдром Леннокса-Гасто характеризуется многочисленных приступов разного типа и умственной отсталостью той или иной степени. У каждого пятого ребёнка, который страдает данной ЭС, ранее был диагностирован синдром Веста.
Дети с данным синдромом могут испытывать различные типы приступов: тонические, миоклонические пароксизмы, атипичные абсансы, атонические пароксизмы и другие. Важной особенностью синдрома является наличие у ребёнка умственной отсталости, которая в начале заболевания может быть незаметна, но после состояние ребёнку ухудшается, если вовремя не поставить диагноз и не начать лечение.
На данный момент существует определенный список препаратов, которые считаются наиболее эффективными для лечения данного синдрома. Подробнее в статье – Синдром Леннокса-Гасто.
Синдром Ландау-Клеффнер (ЛКС, приобретенная афазия) – это редкое заболевание, которое характеризуется эпилептическими приступами с постепенной или внезапной регрессией речевого развития. Болезнь чаще встречается у мальчиков и проявляет себя в возрасте от 3 до 8 лет.
У детей, страдающих данным синдромом, могут наблюдаться различные типы эпилептических приступов, однако особенностью заболевания является регрессия речевого развития. Регрессия при этом может происходить как очень быстро, так и медленно.
Ребёнок начинает постепенно говорить более короткими предложениями, использовать меньшее количество слов, а также перестаёт понимать обращенную к нему речь.
В большинстве случаев, причина появления данного заболевания остаётся неизвестной, однако некоторые исследования зафиксировали, что данное отклонение начинает проявляться у детей с серьезными повреждениями головного мозга в височной доле.
Подробнее на следующей странице – Синдром Ландау-Клеффнера.
- Электрический эпилептический статус медленноволнового сна (ESES)
Электрический эпилептический статус медленноволнового сна (ESES) – это один из синдромов, который относится к эпилептическим энцефалопатиям. ESES является возрастзависимым статусом и считается труднодиагностируемым, так как в ряде случаев не сопровождается характерными клиническими проявлениями. Чаще всего данное отклонение фиксируется у детей в возрасте с 2-х летнего возраста.
ESES проявляется различными типами эпилептических приступов, однако, помимо этого, у пациентов может наблюдаться общая когнитивная регрессия, проблемы с понимаем, регрессия речевых навыков, проблемы с концентрацией внимания, дисфункция органов ротовой полости и другие.
Цель лечения ESES – остановить дальнейшие повреждения головного мозга и ухудшение состояния и когнитивных способностей пациента. Для этого может быть применено как медикаментозное лечение, так и операция.
Лечение эпилептических энцефалопатий
Лечение эпилептический энцефалопатий, в большинстве случаев, агрессивное и может включать в себя как медикаментозное лечение, так и другие методы: назначение строгой диеты и проведение операции. Лечение всегда подбирается индивидуально и зависит не только от вида энцефалопатии, но и от индивидуальных особенностей пациента: сопутствующих заболеваний, степени общей регрессии и т.д.
Прогноз, касательно дальнейшего развития заболевания, всегда будет индивидуальный. Однако, в случае с эпилептическими энцефалопатиями ни в коем случае нельзя затягивать с диагностикой и лечением, ведь прогрессирование заболевания может привести к необратимым последствиям.
Хотите знать, что действительно помогает при эпилепсии и как помочь ребёнку жить максимально полноценной жизнью, несмотря на заболевание?
Раз в неделю мы выпускаем видео или статью о лечении эпилепсии. Это БЕСПЛАТНАЯ и ЕДИНСТВЕННАЯ в своем роде электронная рассылка в мире и мы уверены, что в этих выпусках вы найдёте много полезных рекомендаций для себя и своего ребёнка.
- Можно ли вылечить эпилепсию у детей?
- Переходит ли эпилепсия по наследству?
- Помогают ли альтернативные методы лечения контролировать приступы?
- Что нельзя делать при эпилепсии?
- Сколько должен спать ребёнок с эпилепсией?
- Чем опасны приступы эпилепсии во сне?
- Как помочь пациенту во время приступа?
- Можно ли заниматься спортом?
- Может ли эпилепсия привести к проблемам в учебе, задержке развития, проблем с памятью и поведением?
Введите адрес вашей электронной почты и проверьте почту через 5 минут
***Мы ценим вас и ваше доверие. Наша цель – предоставить вам достоверную информацию о лечении эпилепсии, а также постараться помочь вам или вашим детям жить с этим тяжелым заболеванием. Ни при каких обстоятельствах ваши данные не будут переданы и проданы третьим лицам. Как и вы, мы не любим получать бесполезную почту или рекламу, и постараемся оправдать ваше доверие.
Книга написана простым языком для мам и пап, полна практических советов и рекомендаций эксперта-эпилептолога с мировым именем.
В книге профессора Крамера вы найдёте ответы на многие ваши вопросы об эпилепсии у детей, начиная с видов приступов, правильной диагностики, эффективных методов лечения, и заканчивая практическими советами о том, как повысить качество жизни вашего ребёнка и подготовить его к самостоятельной взрослой жизни.
Читайте также: