
Синдром веста клинические рекомендации
Похожие темы научных работ по клинической медицине , автор научной работы — Акчурина Я.Е., Савинов С.В., Синицына Т.Н., Ситников И.Ю., Данияр Ж.Р.
Особенности течения и лечения эпилептического статуса у детей. Опыт применения внутривенной формы Конвулекса качестве основного средства патогенетического лечения эпилептического статуса
Влияние вальпроатов и синактена-депо на динамику эпилептических приступов и психомоторное развитие у больных синдромом Веста
Раннее назначение внутривенной формы Конвулекса не только улучшает прогноз но и минимализирует осложнения эпилептического статуса у детей раннего возраста с различными формами эпилепсии.
Применение Конвулекса не вызывает угнетения дыхания и гипотензии, что является его неоспоримым преимуществом в сравнении с применением стандартной терапиеи.
Конвулекс в форме раствора для внутривенного введения в терапевтических дозах хорошо переносится и не вызывает серьезных побочных эффектов.
Необходимо отметить, что применение парентеральной формы Конвулекса во время критических состояний, вызванных статусным течением эпилепсии, позволяет после их купирования, обеспечить переход на пероральную форму этого же препарата (Конвулекс в форме капель, сиропа, капсул, таблеток), обеспечивая равновесную концентрацию в крови вальпроевой кислоты и сохраняя достигнутую эффективность в лечении каждого конкретного больного.
В заключение необходимо отметить, что ЭС является одним из наиболее опасных состояний в детской неврологии и нейрохирургии. Своевременное проведение комплекса диагностических и лечебных мероприятий с применение современных препаратов позволяет сохранить жизнь больным эпилепсией, а многих из них, вернуть к полноценной жизни.
1. В.А Карлов. Судорожный и бессудорожный эпилептический статус. Москва, 2007 г.
4. Alfonso I., Perea A., Paez J.C., et al. Intravenous Valproic Acid dose in neonates. Epilepsia, 1999; 40, suppl.7: 143.
5. Campistol J., Fernandes A., Ortega J. Status epilepticus in children. Experience with intravenous valproate. Update of treatment guidelines. - Rev-Neurol., 1999; 24(4): 359-365.
Применение международных протоколов обследования и лечения при ведении пациентов с синдромом Веста
Акчурина Я.Е., Савинов С.В., Синицына Т.Н., Ситников И.Ю., Данияр Ж.Р., Кайруллаев К.К., Смирнова О.Ю., Чулкова И.Ф., Кислякова Е.М., Утебеков Ж.Е. SVS Лаборатория изучения эпилепсии, судорожных состояний и семейного мониторинга имю В.М. Савинова, Алматы, Казахстан
«Самое непостижимое в мире - то, что он
Стремительно развивающаяся медицина постоянно открывает не только новые методы диагностики и лечения, но и раздвигает возможности уже имеющихся.
Прогресс порой, непросто дает вторую жизнь методам исследования, открытым десятки лет назад, а наделяет их новым смыслом!
Прошло чуть больше четверти века и в новом издании профессора Зенкова Л.Р., в трудах К.Ю. Мухина, Е.Д. Белоусовой и многих других мы видим не только развитие методов ЭЭГ диагностики, но и диаметрально противоположный взгляд на синдром Веста.
Частота синдрома Веста в популяции детей с эпилепсией составляет 17—30 %. [4, 11,12]
50-77% случаев имеют своё начало в возрасте от 3 до 7 месяцев. [5]
Синдром Веста представлен возраст зависимой комбинацией эпилептических приступов, ментальных расстройств и гипсаритмии. В случае отсутствия лечения инвалидиза-ция или летальный исход в 100% [5,12] Эти факты говорят об особой актуальности проблемы.
Как и для всех видов эпилепсии, ведущим методом исследования является ЭЭГ [2, 6, 8, 14]
Интериктальным ЭЭГ-коррелятом синдрома Веста является гипсаритмия.
Гипсаритмия является этиопатогенетическим критерием интеллектуального регресса или задержки психомоторного
развития. [2, 5, 7,8, 12,]
Лечение Синдрома Веста и других видов эпилептических энцефалопатий остается до сих пор одной из сложнейших задач. Среди различия методов и медикаментов нет единой, до конца сформированной схемы ведения пациентов. [1, 5, 10, 15, 19]
В последние годы данная проблема получила поступательный импульс. В результате наметились унифицированные пути подхода к началу лечения.
Учитывая агрессивность процесса и отражение этой агрессивности на ЭЭГ в виде гипсаритмии, основной целью лечения является купирование приступов, и устранение гипсаритмии. [12,17]
В момент выявления синдрома Веста рекомендуется немедленно начать лечение вальпроатами.
При отсутствии эффекта от монотерапии в виде санации гипсаритмии на ЭЭГ и купирования приступов рекомендуется подключения следующих препаратов: АКТГ (синактен - депо), Вигабатрин, топирамат, ламотриджин. [10, 11, 12, 15, 17]
Причем, по эффективности, при купировании инфантильных спазмов и гипсаритмии, АКТГ и вигабатрина стоят на первом месте. [1,19]
Хотя и уровень побочных эффектов у этих препаратов тоже высок. [1,10,13,15, 16, 19]
В литературе довольно подробно описаны результаты лечения и эволюция, как клинических проявлений, так и ЭЭГ паттернов. [5,12]
Тем не менее, очень мало практических рекомендаций касающихся протокола нейрофизиологического ведения пациентов с синдромом Веста и другими эпилептическими энцефалопатиями.
основе результатов ЭЭГ контроля.
Второй по важности задачей, мы ставили перед собой анализ влияния ранней диагностики и купирования симптомов заболевания на развитие ребенка.
За период с 5 октября 2004 по 10 января 2010 мы наблюдали 56 пациентов с синдромом Веста.
Из них 38 (67,86%) мальчиков и 18 (32,14%) девочек.
Основными жалобами были приступы в виде инфантильных спазмов, задержка психомоторного развития или регресс в развитии.
Сроки начала заболевания отражены в таблице №1
Таблица № 1 Дебют начала заболевания
Не грубые Грубые
изменения Врожденные структурные
(незначительное пороки изменения
расширение развития (выраженный
Без субарахноидального головного мозга атрофический процесс
видимои пространства, (агенезия или в коре головного
патологи не выраженная гипоплазия мозга, кисты, рубцово
компенсированная мозолистого - атрофические
гидроцефалия, тела, последствия
локальное шизэнцефалия, кровоизлияний,
расширение боковых субкомпенсированные
25% 32,14% 17,86 % 25%
от 2 до 7 месяцев от рождения 45 80,36%
от 7 до 18 месяцев от рождения 9 16,07%
от 18 месяцев и старше 2 3,57%
Не все пациенты обратились за лечением сразу же после возникновения симптомов. Сроки обращаемости и процентное соотношение представлено в таблице №2
сроки обращаемости от момента заболевания абс %
В течении первой недели от начала заболевания 8 14,28
в период со второй недели до месяца от начала заболевания 12 21,43
в период с второго по 12 месяц от начала заболевания 18 32,14
позже года от начала заболевания 18 32,14
Всем пациентам, согласно рекомендации РГМУ и Европейской Ассоциации нейрофизиологов было проведено видео ЭЭГ мониторирование цикла сон - бодрствование. [14]
Если пациент принимал противоэпилептический препарат с нелинейным накоплением в крови, проводилось определение концентрации его в плазме.
Проводилось психоневрологическое тестирование на предмет выявления задержки или регресса развития.
У всех пациентов на ЭЭГ выявлялась гипсаритмия той или иной формы. Результаты ЭЭГ паттернов представлены в таблице №3.
Типичная 10 17,86%
Синхронизированная гипсаритмия 3 5,36%
супресивно взрывной вариант 4 7,14%
ассиметричная (унилатеральная) 11 19,64%
С зональным акцентом 7 12,5%
С региональным акцентом 21 37,5%
Всем пациентам, за исключением трех, проводилось МРТ головного мозга, или КТ. В результате, у 40 пациентов (71,43%) выявились структурные изменения в головном мозге. Структура изменений представлена в таблице № 4.
После постановки диагноза всем пациентам был назначены вальпроат содержащий препарат(конвулекс или депакин).
В результате на субтоксических дозировках у 5 (8,93 %) пациентов отмечались улучшения в клинических проявлениях. Но на ЭЭГ гипсаритмия практически не
В связи с чем, 23 (41.1%) пациентам был начат курс Синактен - депо (АКТГ) [18], 4 (7,14 %) пациентам добавили вигабатрин, 25 (44,64%) родителей пациентов отказались от применения Синактена Депо и вигабатрина в связи с боязнью побочных явлений. Но так как они не отказывались от лечения, им был назначен второй препарат: 18 пациентам топирамат и 7 пациентам ламотриджин.
У 2-х пациентов в момент поступления отмечались выраженные неврологические и соматические отклонения. На МРТ глубокие поражения головного мозга. Несмотря на проводимую терапию пациенты скончались в результате полиорганной патологии.
Еще 2 пациента, отказавшись от традиционного медикаментозного лечения, проводили лечение травами и другими нетрадиционными методами. Данные пациенты скончались в течение года в результате не леченного эпилептического статуса.
Результаты представлены в таблице №5
Как видно из таблицы у 20 пациентов удалось достичь ремиссии. Большая часть положительного результата была достигнута при введении в схему лечения синактен депо или вигабатрина (сабрила).
У всех пациентов, у которых удалось достичь стойкой ремиссии, на МРТ не было грубых структурных изменений. Кроме этого лечение было начато не позже четырех месяцев от начала клинических проявлений заболевания.
1. Применение международных протоколов обследования и лечения пациентов с синдромом Веста позволяет достичь значительных успехов.
2. Результат лечения синдрома Веста зависит от степени органического поражения головного. Чем сохраннее мозг, тем больше шансов на успех в лечении.
3. Степень успеха в лечении зависит от срока обращаемости: наиболее оптимально, если пациенты начинают терапию в сроки от недели до пяти месяцев от начала заболевания. Более позднее обращение приводит к необратимым последствиям.
4. Необходимо обследовать детей не только по протоколу рутинной ээг, но и с применением записи цикла сон бодрствование. Особенно ценно проведение видео ЭЭГ мониторинга.
5. При отсутствии эффекта на максимальных дозировках монотерапии необходимо вводить второй препарат противоэпилептического ряда или АКТГ (синактен депо). При наличии вигабатрина возможно применение в монотерапии с последующим замещением на иные комбинации.
6. Обследование необходимо проводить: каждые десять дней лечения до достижения купирования гипсарит-мии и не менее одного раза в три недели первые полгода ремиссии.
7. При достижении стойкой ремиссии обследование
Стойкая ремиссия (более года) Трансформация в мультифокальную эпилепсию и отсут. ремиссии Трансформация в синром Леннокса - Гасто Не известный исход Летальный исход
Терапия включающая синактен депо 13 3 5 2
Терапия включающая вигабатрин (сабрил) 5 2 1
Лечение моно и поли терапией без снактен депо или вигабатрина (сабрила) 2 12 8 4 1
Пациенты оказавшиеся от лечения или поступившие в терминальной стадии 0 0 0 4
необходимо проводить один раз в четыре месяца в виде ночного мониторинга и лучше видео ЭЭГ.
8. Для успешного лечения крайне необходимо проведение психологического тренинга с родителями и организация круглосуточной горячей линии. Данные мероприятия повысят ответственность и понимание родителей проблемы заболевания, а так же поможет избежать или снизить осложнения от терапии.
1. Белоусов Ю.Б., Моисеев В.С., Лепахин В.К. "Клиническая фармакология и фармакотерапия". Руководство для врачей. М: "Униерсум Паблишинг", 2000 г
2. Благосклонова Н.К., Новикова Л.А. "Детская клиническая электроэнцефалография". Руководство для врачей. М: "Медицина", 1994 г. - 255 с.
3. Броун Т., Холмс Г. "Эпилепсия. Клиническое руководство" Пер. с англ. М: "Издательство БИНОМ", 2006 г. - 288 с.
4. Гузева В.И. "Эпилепсия и неэпилептические пароксизмальные состояния у детей", М: "Медицинское информационное агенс-тво", 2007 год.
5. Евтушенко С.К, Омельяненко А.А. "Клиническая электроэнцефалография у детей" Донецк 2005
6. Жирмунская Е.А. "Клиническая электроэнцефалография"
(цифры, гистограммы, иллюстрации). М: отпечатано в типографии ТОО "Вега-Принт", 1993 г.
7. Зенков Л.Р. Ронкин М.А. Функциональная диагностика нервных болезней Москва, Медицина 1982 стр120.
8. Зенков Л.Р."Клиническая электроэнцефалография" (с элементами эпилептологии). Руководство для врачей. М: "МЕДпресс-информ", 2004 г - 368 с.
Л.Р."Непароксизмальные эпилептические расстройства" (Руководство для врачей). М: "МЕДпресс-информ", 2007 г. - 280 с.
10. Зенков Л.Р., Притыко А.Г. "Фармакорезистентные эпилепсии" М.:"МЕДпресс-ин-форм" 2003 - 207с.
11. Мухин К.Ю., Петрухин
А.С."Эпилептические синдромы. Диагностика и стандарты терапии" Справочное руководство" М.: 2005 г.
12. Мухин К.Ю., Петрухин А.С., Глухова Л.Ю. "Эпилепсия. Атлас электро-клинической диагностики". М: "Альварес Паблишинг", 2004 г. - 440 с.
13. Справочник ВИДАЛЬ. Лекарственные препараты в Казахстане. М: Астра Фарм Сервис, 2008 г. - 848 с.
14. Строганова Т.А., Дегтярева М.Г., Володин Н.Н. "Электроэнцефалография в неонатологии" руководство для врачей. М.:"ГЭОТАР-Медиа" 2005 - с 279
15. Темин П.А., Никонорова М.Ю., "Эпилепсии и судорожные синдромы у детей". Руководство для врачей. 1999 г. - 656 с.
16. Темин П.А., Ермаков А.Ю., Белоусова Е.Д. Лечение инфантильных спазмов Российский вестник перинатологии и педиатрии, N3-1999, с.23-30
18. M. Ito, "Low-dose synthetic ACTH therapy for West syndrome: initial effects and long-term outcome" Japan Medical Center for Children, Moriyama 2004
Диагностика и лечение препаратом ламиктал различных форм
эпилепсии у детей
Лепесова М.М., Казакенова А.К., Текебаева Л.А., Исабекова А.А., Мырзалиева Б.Д., Джаксыбаева А.Х. Алматинский Государственный институт усовершенствования врачей, Алматы, Казахстан
Эпилепсия является распространенным, хроническим и дезадаптирующим детей заболеванием, которым страдает около 50 миллионов человек во всем мире. Результаты эпидемиологических исследований свидетельствуют, что эпилепсия развивается у 40-70 больных на 100 000 населения в течение года (каждый год диагностируется 100 000 новых случаев). В настоящее время частота заболеваемости эпилепсией сравнивается с распространенностью сахарного диабета 1 типа.
Наличие эпилепсии связано не только с инвалидиза-цией и снижением качества жизни больных, но и с повышенной летальностью. Все большее внимание в последние годы привлекает так называемая «внезапная, неожиданная смерть при эпилепсии (англ.- «sudden unexpected death in
При своевременной и адекватной терапии свыше, чем у 2/3 больных эпилепсией удается избавиться от припадков и восстановить качество жизни. При лечении детей с эпилепсией основной целью является контроль припадков. Немаловажным при эпилепсии являются такие показатели, как когнитивные нарушения, способность к самообслуживанию и учебе. Значение также имеет и минимизация побочных эффектов противоэпилептической терапии со снижением риска возникновения эпилептического статуса или внезапной смерти больных.
Подходы к терапии разнятся у больных с первично выявленным заболеванием и у пациентов с рефракторной к
Синдром Веста – эпилептический синдром, характеризующийся триадой симптомов: инфантильными спазмами, гипсаритмией на межприступной ЭЭГ, регрессом или задержкой психомоторного развития. Согласно современным представлениям, для постановки диагноза достаточно наличие двух из трех критериев. Частота встречаемости составляет 1 на 2000 новорожденных. Cтатья представляет собой обзор современных представлений об этиологии, патогенезе, клинических проявлениях, диагностике и лечении синдрома Веста. Отдельный раздел статьи иллюстрирует сложности трактовки приступной и межприступной энцефалограммы при инфантильных спазмах. В настоящее время, кроме классической гипсаритмии, описывается пять вариантов модифицированной гипсаритмии: синхронизированный вариант, асимметричный вариант, с устойчивым фокусом спайков или острых волн, с эпизодами уплощения ритмики, с высокоамплитудной асинхронной медленной активностью. Лечение, вне зависимости от этиологии, рекомендовано начинать с гормональной терапии, при отсутствии эффекта препаратом выбора является вигабатрин; обсуждается тактика его применения.
Ключевые слова
Об авторах
Адрес: ул. Островитянова, д. 1, Москва, Россия, 11799
Адрес: ул. Островитянова, д. 1, Москва, Россия, 117997
Адрес: ул. Островитянова, д. 1, Москва, Россия, 117997
Адрес: Ленинский пр-т, 117, Москва, Россия, Р 119571
Адрес: Ленинский пр-т, 117, Москва, Россия, Р 119571
Адрес: ул. Островитянова, д. 1, Москва, Россия, 117997
Список литературы
1. Auvin S., Cilio M. R., Vezzani A. Current understanding and neurobiology of epileptic encephalopathies. DOI: 10.1016/j.nbd.2016.03.007.
3. Мухин К. Ю., Миронов М. Б. Эпилептические спазмы. Русский журнал детской неврологии. 2014; 9 (4): 20-29. DOI:10.17650/2073-8803-2014-9-4-20-29.
4. Iype M., Puthuvathra Abdul Mohammed Kunju, Geetha Saradakutty, Devi Mohan, and Shahanaz Ahamed Mohammed Khan. The early electroclinical manifestations of infantile spasms: A video EEG study. Ann Indian Acad Neurol. 2016 Jan-Mar; 19 (1): 52-57.
5. Iype M., Kunju P. A., Saradakutty G., Mohan D., Khan S. A. The early electroclinical manifestations of infantile spasms: A video EEG study. Ann Indian Acad Neurol. 2016 Jan- Mar; 19 (1): 52-7. DOI: 10.4103/0972-2327.168627.
6. Howell K. B., Harvey A. S., Archer J. S. Epileptic encephalopathy: use and misuse of a clinically and conceptually important concept. Epilepsia. 2016; 57 (3): 343-7.
7. Pavone P., Striano P., Falsaperla R., Pavone L., Ruggieri M. Management of infantile spasms. Transl Pediatr. 2015 Oct; 4 (4): 260-70. DOI: 10.3978/j.issn.2224-4336.2015.09.01.
8. Kodera H., Ohba C., Kato M., Maeda T., Araki K., Tajima D., Matsuo M., Hino-Fukuyo N., Kohashi K., Ishiyama A., Takeshita S., Motoi H., Kitamura T., Kikuchi A., Tsurusaki Y., Nakashima M., Miyake N., Sasaki M., Kure S. Haginoya K., Saitsu H., Matsumoto N. De novo GABRA1 mutations in Ohtahara and West syndromes. Epilepsia. 2016 Apr; 57 (4): 566-73. DOI: 10.1111/epi.13344.
9. Taghdiri M. M., Nemati H. Infantile spasm: a review article. Iran J Child Neurol. 2014 Summer; 8 (3): 1-5.
10. Munakata M., Togashi N., Sakamoto O., Haginoya K., Kobayashi Y., Onuma A., Iinuma K., Kure S. Reduction in glutamine/glutamate levels in the cerebral cortex after adrenocorticotropic hormone therapy in patients with west syndrome. Tohoku J Exp Med. 2014; 232 (4): 277-83.
11. Feng S., Ma S., Jia C., Su Y., Yang S., Zhou K., Liu Y., Cheng J., Lu D., Fan L., Wang Y. Sonic hedgehog is a regulator of extracellular glutamate levels and epilepsy. EMBO Rep. 2016 May; 17 (5): 682-94. DOI: 10.15252/embr.201541569.
12. Frost JD Jr1, Hrachovy R,A. Pathogenesis of infantile spasms: a model based on developmental desynchronization. J Clin Neurophysiol. 2005 Jan-Feb;22(1):25-36.
13. Li- Rong Shao, Carl E Stafstrom. Pediatric epileptic encephalopathies. Pathophysiology and animal models. 2016; 5 (4): 2-12.
14. Elaine C. Wirrell, Renee A., Shellhaas, Charuta Joshi, Cynthia Keator, Shilpi Kumar, Wendy G. How should children with West syndrome be efficiently and accurately investigated? Results from the National Infantile Spasms Consortium. Mitchell, and Pediatric Epilepsy Research Consortium (PERC). Epilepsia. 2015; 56 (4): 617-625. DOI: 10.1111/epi.12951.
15. d’Orsi G., Demaio V., Minervini M. G. Adult epileptic spasms: a clinical and video-polygraphic study. Epileptic Disord. 2007 Sep; 9 (3): 276-83. Epub 2007 Sep 20.
16. Scheffer I. E., Berkovic S., Capovilla G., et al. ILAE classification of the epilepsies. Position paper of the ILAE Commission for Classification and Terminology. Epilepsia (in press 2017).
18. Yilmaz S., Tekgul H., Serdaroglu G., Akcay A., Gokben S. Evaluation of ten prognostic factors affecting the outcome of West syndrome. Acta Neurol Belg. 2016 Dec; 116 (4): 519-527. Epub 2016 Feb 5.
19. Xue J., Qian P., Li H., Yang H., Liu X., Zhang Y., Yang Z. Atonic elements combined or uncombined with epileptic spasms in infantile spasms. Clin Neurophysiol. 2017 Jan; 128 (1): 220-226. DOI: 10.1016/j.clinph.2016.11.008. Epub 2016 Nov 20.
20. Gurkas E., et al. Sleep-Wake Distribution and Circadian Patterns of Epileptic Seizures in Children. Eur J Paediatr Neurol. 2016 Apr 13; 20 (4): 549-554.
21. Pavone P., Striano P., Falsaperla R., Pavone L., Ruggieri M. Infantile spasms syndrome, West syndrome and related phenotypes: what we know in 2013. Brain Dev. 2014 Oct; 36 (9): 739-51. doi: 10.1016/j.braindev.2013.10.008.
22. Wilmshurst J. M., Roland C. Ibekwe and Finbar J. K. O’Callaghan. Epileptic spasms – 175 years on: Trying to teach an old dog new tricks. Sunday, January 1, 2017.
23. Gul Mert G., Herguner M. O., Incecik F., Altunbasak S., Sahan D., Unal I. Risk factors affecting prognosis in infantile spasm. Int J Neurosci. 2017 Mar 29; 1-7. DOI: 10.1080/00207454.2017.1289379.
24. Айкарди Жан. Заболевания нервной системы у детей. 2013; 2: 660-667.
25. Teresa Rand`o, Adina Bancale, Giovanni Baranello, Margherita Bini, et al. Visual Function in Infants with West Syndrome: Correlation with EEG Patterns. Epilepsia. 2004; 45 (7): 781–786.
26. Baranello G., Rando T., Bancale A., et al. Auditory attention at the onset of West syndrome: Correlation with EEG patterns and visual function Brain & Development. 2006; 28: 293-299.
27. Gastaut H., Roger J., Soulayrol R., Salamon G., Regis H., Lob H. Infantile myoclonic encephalopathy with hypsarrhythmia (West›s syndrome) and Bourneville›s tuberous sclerosis. J Neurol Sci. 1965 Mar – Apr;2(2):140-60.
28. Hrachovy R. A., Frost JD Jr, Kellaway P. Hypsarrhythmia: variations on the theme. Epilepsia. 1984 Jun;25(3):317-25.
29. Мухин К. Ю., Петрухин А. С., Холин А. А. Эпилептические энцефалопатии и схожие синдромы у детей. М Арт.Сервис.Лтд.; Санкт-Петербург ; 2011 г. 680 стр
30. Айвазян С. О. Эволюция припадков и ЭЭГ характеристик при ранних детских формах эпилепсии. Автореф. дисс. … канд. мед. наук. М. 1999.
31. Pramote Laoprasert. Atlas of Pediatric EEG. 2011.
34. Wheless J. W., Clarke D. F., Arzimanoglou A., Carpenter D. Treatment of pediatric epilepsy: European expert opinion, 2007. Epileptic Disord. 2007; 9: 353-412.
35. Белоусова Е. Д., Шулякова И. В., Охапкина Т. Г. Гормональная терапия синдрома Веста. Журнал неврологии и психиатрии им. C. C. Корсакова. 2016. Т. 116. № 9-2. С. 61- 66. DOI: 10.17116/jnevro20161169261-66
36. Белоусова Е. Д., Дорофеева М. Ю., Охапкина Т. Г. Лечение эпилепсии при туберозном склерозе. Эпилепсия и пароксизмальные состояния. 2016; 2: 37-42.
37. Moavero R., Marciano S., Graziola F., Curatolo P. Combined targeted treatment in early onset epilepsy associated with tuberous sclerosis. Epilepsy Behav Case Rep. 2016 Jan 13; 5: 13-6. DOI: 10.1016/j.ebcr.2015.12.001.
38. Kayani S., Sirsi D. The safety and tolerability of newer antiepileptic drugs in children and adolescents. J Cent Nerv Syst Dis. 2012 Mar 8;4:51-63. doi: 10.4137/JCNSD.S509
40. Riikonen R., Rener-Primec Z., Carmant L., Dorofeeva M., Hollody K., Szabo I., Krajnc B. S., Wohlrab G., Sorri I. Does vigabatrin treatment for infantile spasms cause visual field defects? An international multicentre study. Dev Med Child Neurol. 2015 Jan; 57 (1): 60-7.
41. Origlieri C., Geddie B., Karwoski B., Berl M. M., Elling N., McClintock W., Alexander J., Bazemore M., de Beaufort H., Hutcheson K., Miller M., Taylormoore J., Jaafar M. S., Madigan W. Optical coherence tomography to monitor vigabatrin toxicity in children. J AAPOS. 2016 Apr; 20 (2): 136-40. DOI: 10.1016/j.jaapos.2015.10.020.
42. Fernandez-Garcia M. A., Garcia-Penas J. J., Gomez-Martin H., Perez-Sebastian I., Garcia- Esparza E., Sirvent-Cerda S. Reversible alterations in the neuroimages associated with vigabatrine treatment in infants with epileptic spasms. Rev Neurol. 2017 Feb 16; 64 (4): 169-174.
43. O’Callaghan F. J., Edwards S. W., Alber F. D., Hancock E., Johnson A. L., Kennedy C. R., Likeman M., Lux A. L., Mackay M., Mallick A. A., Newton R. W., Nolan M., Pressler R., Rating D., Schmitt B., Verity C. M., Osborne J. P. Safety and effectiveness of hormonal treatment versus hormonal treatment with vigabatrin for infantile spasms (ICISS): a randomised, multicentre, open-label trial. Lancet Neurol. 2017 Jan; 16 (1): 33-42. doi: 10.1016/S1474-4422(16)30294-0. Epub 2016 Nov 10.
44. Bachman D. S. Use of valproic acid in treatment of infantile spasms. Arch Neurol. 1982 Jan; 39 (1): 49-52.
45. Rajaraman R. R., Lay J., Alayari A., Anderson K., Sankar R., Hussain S. A. Prevention of infantile spasms relapse: Zonisamide and topiramate provide no benefit. Epilepsia. 2016 Aug; 57 (8): 1280-7. DOI: 10.1111/epi.13442. Epub 2016 Jun 17.
46. Song J. M., Hahn J., Kim S. H., Chang M. J. Efficacy of Treatments for Infantile Spasms: A Systematic Review. Clin Neuropharmacol. 2017 Mar/Apr; 40 (2): 63-84. DOI: 10.1097/WNF.0000000000000200.
47. Riikonen R., Mankinen K., Gaily E. Long-term outcome in pyridoxine – esponsive infantile epilepsy. Eur J Paediatr Neurol. 2015 Nov; 19 (6): 647-51. DOI: 10.1016/j.ejpn.2015.08.001.
48. Wilmshurst J. M., Ibekwe R. C., O’Callaghan F. J. Epileptic spasms – 175 years on: Trying to teach an old dog new tricks. Seizure. Seizure. 2017 Jan;44:81-86. doi: 10.1016/j.seizure.2016.11.021
49. Altunel A., Sever A., Altunel E. Ц. Hypsarrhythmia paroxysm index: A tool for early prediction of infantile spasms. Epilepsy Res. 2015 Mar; 111: 54-60. doi: 10.1016/j.eplepsyres.2015.01.005.
Генерализованная эпилепсия (ГЭ) – хроническое заболевание головного мозга, характеризующиеся повторными приступами с нарушением двигательных, чувствительных, вегетативных, мыслительных или психических функций, возникающими вследствие чрезмерных нейронных разрядов в обоих полушариях головного мозга.
ГЭ – является единым заболеванием, представляющим отдельные формы с электро-клиническими особенностями, подходом к лечению и прогнозом.
Код протокола: H-P-003 "Генерализованная эпилепсия у детей, острый период"
Для стационаров педиатрического профиля
Классификация
Согласно Международной классификации 1989 г. (Международная лига борьбы с эпилепсией) в основу генерализованной эпилепсии положена генерализованность эпилептической активности.
Внутри ГЭ выделяют формы : идиопатические, симптоматические и криптогенные.
Генерализованные виды эпилепсии и синдромы:
1. Идиопатические (с возраст-зависимым дебютом). МКБ-10: G40.3:
- доброкачественные семейные неонатальные судороги;
- доброкачественные идиопатические неонатальные судороги;
- доброкачественная миоклоническая эпилепсия раннего детского возраста;
- детская абсансная эпилепсия (МКБ-10: G40.3);
- ювенильная абсансная эпилепсия;
- ювенильная миоклоническая эпилепсия;
- эпилепсия с приступами пробуждения;
- другие виды идиопатической генерализованной эпилепсии (МКБ-10: G40.4);
- эпилепсия с приступами, провоцируемыми специфическими факторами.
2. Криптогенные и (или) симптоматические (с возраст-зависимым дебютом) - МКБ-10: G40.5:
- синдром Веста (инфантильные спазмы);
- синдром Леннокса-Гасто;
- эпилепсия с миоклонически-астатическими приступами;
- эпилепсия с миоклоническими абсансами.
3. Симптоматические.
3.2 Специфические синдромы.
Диагностика
- на наличие в анамнезе неонатальных приступов, судорог при повышении температуры (являются факторами риска развития эпилепсии);
- токсические, ишемические, гипоксические, травматические и инфекционные поражения мозга, включая внутриутробный период (могут быть причинами данного заболевания).
Физикальное обследование:
- наличие судорог;
- характер приступов;
- семейная предрасположенность;
- возраст дебюта;
- длительность приступа.
Лабораторные исследования
Количество лейкоцитов и тромбоцитов определяют для исключения фолиево-дефицитной анемии и связанных с этим вторичных изменений костного мозга, что клинически проявляется снижением уровня лейкоцитов и тромбоцитов;
Снижение удельного веса мочи может свидетельствовать о появлении почечной недостаточности, что требует уточнения дозировки препаратов и тактики лечения.
Дифференциальный диагноз: нет.
Лечение
Первый врач, заставший эпилептический припадок, должен подробно его описать, включая признаки, которые предшествовали припадку и возникали после его окончания.
Больных необходимо направлять на полное неврологическое обследование для подтверждения диагноза и выяснения этиологии.
Лечение эпилепсии начинают только после установления точного диагноза. По мнению большинства специалистов, лечение эпилепсии следует начинать после повторного приступа.
Немедикаментозное лечение: необходим полноценный ночной сон.
Медикаментозное лечение
Лечение эпилепсии должно осуществляться в зависимости от формы эпилепсии, а затем от характера приступов – с базового препарата для данной формы эпилепсии. Стартовая доза составляет примерно 1/4 от средней терапевтической. При хорошей переносимости препарата дозировка увеличивается примерно до 3/4 от средней терапевтической дозы в течение 2-3 недель.
При отсутствии или недостаточном эффекте доза повышается до средней терапевтической.
При отсутствии эффекта от терапевтической дозы в течение 1 месяца необходимо дальнейшее постепенное увеличение дозы до получения выраженного положительного эффекта или появления побочных эффектов.
При отсутствии терапевтического эффекта и появлении признаков интоксикации, препарат постепенно заменяется на другой.
При получении выраженного терапевтического эффекта и наличии побочных эффектов, необходимо оценить характер и степень выраженности последних, затем решить вопрос о продолжении лечения или замене препарата.
Замена барбитуратов и бензодиазепинов должна производиться постепенно в течение 2-4-х недель и более ввиду наличия выраженного синдрома отмены. Замена других антиэпилептических препаратов (АЭП) может быть осуществлена более быстро – за 1-2 недели. Оценка эффективности препарата может быть произведена лишь не ранее 1 месяца с момента начала его приема.
Синдром Веста – это возрастзависимый эпилептический синдром, относящийся к группе младенческих эпилептических энцефалопатий и характеризующийся особым видом приступов – инфантильными спазмами.
Синдром впервые был описан в 1841 году английским доктором Вестом, когда данное состояние наблюдалось у его ребёнка.
Есть несколько видов синдрома Веста: симптоматический, криптогенный и идиопатический. У каждого вида есть свои характерные ему особенности, течение заболевания и свой прогноз на будущее.
При данном виде заболевания очень важно провести раннюю диагностику, определить диагноз и начать соответствующее лечение, чтобы предотвратить тяжелые когнитивные нарушения. В большинстве случаев, заболевание проявляется у детей в возрасте нескольких месяцев.
Симптомы синдрома Веста
Как правило, синдром проявляется в первые месяцы жизни или в течение первого года жизни ребёнка (до года).
Основным симптомом заболевания являются приступы в виде спазмов. Во время приступа у ребёнка сокращаются различные группы мышц тела и конечностей. Приступы носят серийный характер и, в большинстве случаев, длятся до 15 минут.
Инфантильные спазмы всегда начинаются непредсказуемо и могут возникнуть в любое время суток, но обычно наблюдаются во время пробуждения или засыпания ребёнка. В некоторые случаях, могут наблюдаться генерализованные сокращения мышц, но наиболее частыми являются локальные спазмы.

Причины синдрома Веста
В большинстве случаев, синдром носит симптоматический характер и его причиной является поражение головного мозга по какой-либо причине. Самой распространённой причиной заболевания у детей является гипоксия при родах. Гипоксия – это кислородное голодание головного мозга, которое может возникнуть из-за патологических состояний, при неблагоприятном течении беременности или осложнениях при родах, например, обвитие пуповиной шеи плода.
Помимо этого, к заболеванию могут привести:
- инфекционные заболевания головного мозга в период после рождения;
- структурные патологии головного мозга;
- туберозный склероз;
- генетические и метаболические нарушения;
- структурные изменения в головном мозге;
- ряд других причин.
Внимание родителей! Если причина появления синдрома у вашего ребёнка не обнаружена, крайне важно провести генетические тесты перед планированием следующего ребёнка.
Диагностика
- ЭЭГ – данное исследование крайне важно во время диагностики синдрома Веста, так как на электроэнцефалограмме можно будет заметить определенную, специфическую для заболевания картину.
- МРТ – необходимо для определения наличия поражений головного мозга. В маленьком возрасте данное исследование проводится под наркозом.
- Генетические тесты и метаболические исследования – помогут дать больше информации о вашем случае и подтолкнуть к более эффективному протоколу лечения.
- Дополнительные проверки – они могут быть назначены при необходимости исключения наличия иных заболеваний (о них мы не будет говорить в данной статье, так как это достаточно объемная тема).
ВАЖНО! При первых появлениях настораживающих или подозрительных движений у младенца, следует как можно скорее проконсультироваться с педиатром и неврологом. Обратите внимание, если у малыша наблюдаются регулярные, серийные, цикличные движения. Желательно не ждать очереди даже пару недель!
Иногда родители обращаются за помощью не сразу, особенно если инфантильные спазмы не происходили сериями, что приводит к необративным последствиям.
Лечение синдрома Веста
В большинстве случаев, синдром Веста у детей требуется агрессивное медикаментозное лечение, чтобы предотвратить дальнейшие когнитивные нарушения у ребенка.
Наиболее эффективными противоэпилептическими препаратами при синдроме Веста считаются Сабрил и стероиды в больших дозировках.
Как показывает опыт наших иностранных пациентов, во многих странах мира, в том числе в России, Украине, Казахстане и других странах бывшего СССР, клинические рекомендации могут не включать достаточно высокие дозировки из-за тяжелых побочных эффектов, что, к сожалению, может не дать нужного терапевтического эффекта.
Прогноз
К нам часто поступает вопрос: можно ло ли вылечить синдром Веста и каков прогноз на будущее? Смотрите статистику и факты.
- Примерно у половины детей эпилептические приступы прекратятся.
- В плане моторики у большинства детей не будет проблемы с развитием.
- Симптоматический и криптогенный виды синдрома Веста часто сопровождаются определенной задержкой когнитивного развитии, причем во многих случаях на ранних стадиях тяжело определить тяжесть поражения или его наличие, что затрудняет прогноз.
- У каждого пятого ребёнка с синдромом Веста после наблюдается синдром Леннокса-Гасто.
- В большинстве случаев, дети с данным синдромом страдают от фармакорезистентной эпилепсии, устойчивой к медикаментозной терапии.
Хотите узнать больше о синдроме Веста и необходимом вашему ребёнку лечении?
Подробную информацию о синдроме Веста, диагностике и протоколах лечения можно найти в книге профессора Ури Крамера “Детская эпилепсия от А до Я”. Профессор даёт четкие рекомендации родителям на что нужно обратить внимание на том или ином этапе лечения, а также даёт прогноз развития заболевания в будущем.
Хотите знать, что действительно помогает при эпилепсии и как помочь ребёнку жить максимально полноценной жизнью, несмотря на заболевание?
Раз в неделю мы выпускаем видео или статью о лечении эпилепсии. Это БЕСПЛАТНАЯ и ЕДИНСТВЕННАЯ в своем роде электронная рассылка в мире и мы уверены, что в этих выпусках вы найдёте много полезных рекомендаций для себя и своего ребёнка.
- Можно ли вылечить эпилепсию у детей?
- Переходит ли эпилепсия по наследству?
- Помогают ли альтернативные методы лечения контролировать приступы?
- Что нельзя делать при эпилепсии?
- Сколько должен спать ребёнок с эпилепсией?
- Чем опасны приступы эпилепсии во сне?
- Как помочь пациенту во время приступа?
- Можно ли заниматься спортом?
- Может ли эпилепсия привести к проблемам в учебе, задержке развития, проблем с памятью и поведением?
Введите адрес вашей электронной почты и проверьте почту через 5 минут
***Мы ценим вас и ваше доверие. Наша цель – предоставить вам достоверную информацию о лечении эпилепсии, а также постараться помочь вам или вашим детям жить с этим тяжелым заболеванием. Ни при каких обстоятельствах ваши данные не будут переданы и проданы третьим лицам. Как и вы, мы не любим получать бесполезную почту или рекламу, и постараемся оправдать ваше доверие.

Книга написана простым языком для мам и пап, полна практических советов и рекомендаций эксперта-эпилептолога с мировым именем.
В книге профессора Крамера вы найдёте ответы на многие ваши вопросы об эпилепсии у детей, начиная с видов приступов, правильной диагностики, эффективных методов лечения, и заканчивая практическими советами о том, как повысить качество жизни вашего ребёнка и подготовить его к самостоятельной взрослой жизни.
Эпилептическая энцефалопатия – это вид эпилепсии, проявления которой могут привести к повреждению головного мозга.
Эпилептическая энцефалопатия является тяжелым вариантом течения заболевания, при котором, в большинстве случаев, наблюдаются не только эпилептические приступы, но и когнитивные нарушения различной степени тяжести.
Дебютируют данные заболевания, в большинстве случаев, в раннем возрасте на фоне нормального или уже имеющегося нарушенного развития. Медицине известно несколько десятков различных ЭЭ, с наиболее часто встречающимися из них мы познакомим вас в данной статье.
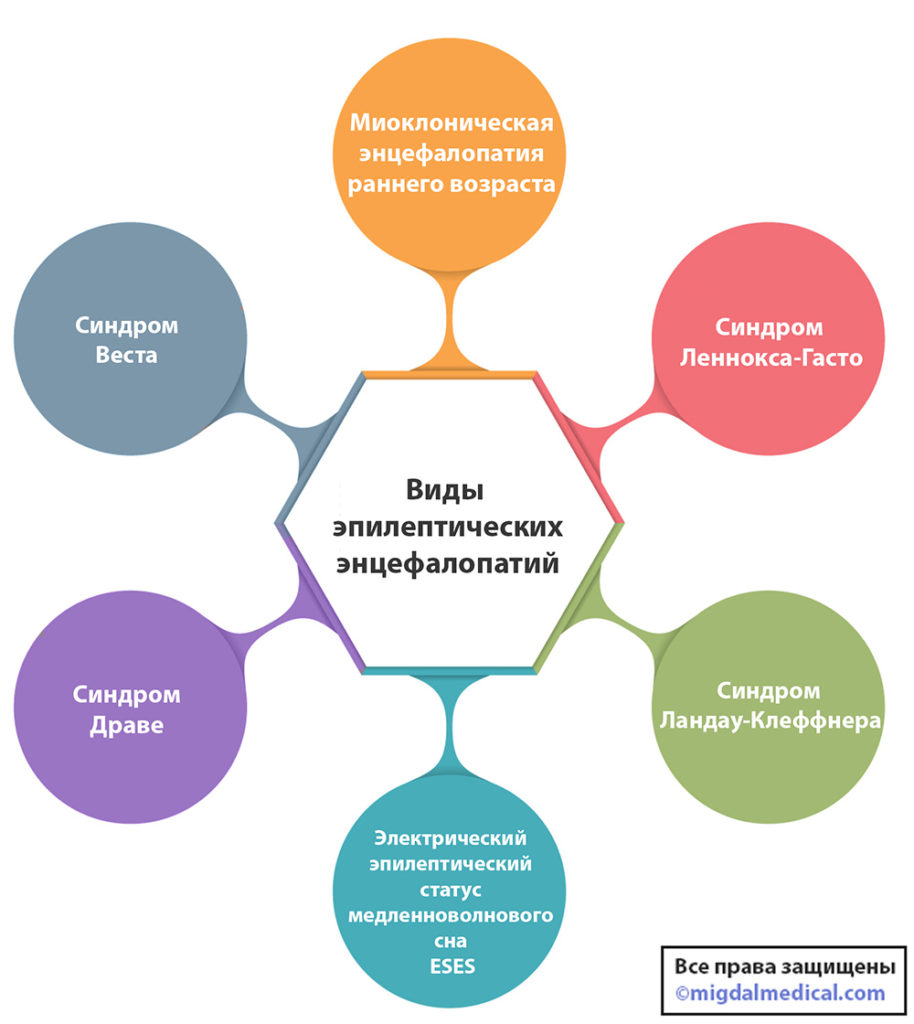
Виды эпилептических энцефалопатий
- Миоклоническая энцефалопатия раннего возраста
Ранняя миоклоническая энцефалопатия является редкой возраст-зависимой формой эпилепсии. Диагноз может быть установлен посредством ЭЭГ, так как на нём проявляется специфическая эпилептическая активность головного мозга, свойственная именно данной ЭЭ. К сожалению, данный тип болезни, в большинстве случаев, является устойчивым к медикаментозному лечению противоэпилептическими препаратами.
Синдром Веста – это возрастзависимый эпилептический синдром, относящийся к группе младенческих эпилептических энцефалопатий и характеризующийся особым видом приступов – инфантильными спазмами. Данное заболевание может привести к серьезным когнитивным нарушениям, а потому крайне важно как можно раньше провести диагностику и назначить подходящее пациенту лечение.
Причиной синдрома Веста является поражение головного мозга, которое наиболее часто появляется во время гипоксии (кислородного голодания головного мозга) при родах. Также причиной возникновения повреждений могут стать инфекционные заболевания, структурные патологии мозга, генетические нарушения и другие.
В большинстве случаев, синдром Веста у детей требуется агрессивное медикаментозное лечение, чтобы предотвратить дальнейшие когнитивные нарушения у ребенка. Узнать больше о синдроме и его лечении вы можете на следующей странице – Синдром Веста.
Синдром Драве – это детская эпилептическая энцефалопатия наследственного характера, которая проявляется эпилептиформными приступами и приводит к когнитивным нарушениям. От данного заболевания чаще страдают девочки, нежели мальчики. Как правило, до 5 месяцев родители не замечают каких-либо отклонений в развитии ребёнка и у него ответствуют судороги, однако после 5 месяцев энцефалопатия даёт о себе знать.
Во время синдрома у ребёнка наблюдается большое количество разнообразных приступов: фокальные, фебрильные, миоклонические и генерализованные судорожные припадки. Помимо этого, у детей проявляются другие синдромы: проблемы с моторикой, тремор, дизартрия, атаксия и другие.
К сожалению, большинство детей, страдающих данным синдромом, не восприимчивы к медикаментозной терапии. Однако, в некоторых случаях удаётся достичь снижения количества приступов благодаря противоэпилептическим препаратам.
Больше узнать о данном синдроме можно на следующей странице – Синдром Драве.
Синдром Леннокса-Гасто характеризуется многочисленных приступов разного типа и умственной отсталостью той или иной степени. У каждого пятого ребёнка, который страдает данной ЭС, ранее был диагностирован синдром Веста.
Дети с данным синдромом могут испытывать различные типы приступов: тонические, миоклонические пароксизмы, атипичные абсансы, атонические пароксизмы и другие. Важной особенностью синдрома является наличие у ребёнка умственной отсталости, которая в начале заболевания может быть незаметна, но после состояние ребёнку ухудшается, если вовремя не поставить диагноз и не начать лечение.
На данный момент существует определенный список препаратов, которые считаются наиболее эффективными для лечения данного синдрома. Подробнее в статье – Синдром Леннокса-Гасто.
Синдром Ландау-Клеффнер (ЛКС, приобретенная афазия) – это редкое заболевание, которое характеризуется эпилептическими приступами с постепенной или внезапной регрессией речевого развития. Болезнь чаще встречается у мальчиков и проявляет себя в возрасте от 3 до 8 лет.
У детей, страдающих данным синдромом, могут наблюдаться различные типы эпилептических приступов, однако особенностью заболевания является регрессия речевого развития. Регрессия при этом может происходить как очень быстро, так и медленно.
Ребёнок начинает постепенно говорить более короткими предложениями, использовать меньшее количество слов, а также перестаёт понимать обращенную к нему речь.
В большинстве случаев, причина появления данного заболевания остаётся неизвестной, однако некоторые исследования зафиксировали, что данное отклонение начинает проявляться у детей с серьезными повреждениями головного мозга в височной доле.
Подробнее на следующей странице – Синдром Ландау-Клеффнера.
- Электрический эпилептический статус медленноволнового сна (ESES)
Электрический эпилептический статус медленноволнового сна (ESES) – это один из синдромов, который относится к эпилептическим энцефалопатиям. ESES является возрастзависимым статусом и считается труднодиагностируемым, так как в ряде случаев не сопровождается характерными клиническими проявлениями. Чаще всего данное отклонение фиксируется у детей в возрасте с 2-х летнего возраста.
ESES проявляется различными типами эпилептических приступов, однако, помимо этого, у пациентов может наблюдаться общая когнитивная регрессия, проблемы с понимаем, регрессия речевых навыков, проблемы с концентрацией внимания, дисфункция органов ротовой полости и другие.
Цель лечения ESES – остановить дальнейшие повреждения головного мозга и ухудшение состояния и когнитивных способностей пациента. Для этого может быть применено как медикаментозное лечение, так и операция.
Лечение эпилептических энцефалопатий
Лечение эпилептический энцефалопатий, в большинстве случаев, агрессивное и может включать в себя как медикаментозное лечение, так и другие методы: назначение строгой диеты и проведение операции. Лечение всегда подбирается индивидуально и зависит не только от вида энцефалопатии, но и от индивидуальных особенностей пациента: сопутствующих заболеваний, степени общей регрессии и т.д.
Прогноз, касательно дальнейшего развития заболевания, всегда будет индивидуальный. Однако, в случае с эпилептическими энцефалопатиями ни в коем случае нельзя затягивать с диагностикой и лечением, ведь прогрессирование заболевания может привести к необратимым последствиям.
Хотите знать, что действительно помогает при эпилепсии и как помочь ребёнку жить максимально полноценной жизнью, несмотря на заболевание?
Раз в неделю мы выпускаем видео или статью о лечении эпилепсии. Это БЕСПЛАТНАЯ и ЕДИНСТВЕННАЯ в своем роде электронная рассылка в мире и мы уверены, что в этих выпусках вы найдёте много полезных рекомендаций для себя и своего ребёнка.
- Можно ли вылечить эпилепсию у детей?
- Переходит ли эпилепсия по наследству?
- Помогают ли альтернативные методы лечения контролировать приступы?
- Что нельзя делать при эпилепсии?
- Сколько должен спать ребёнок с эпилепсией?
- Чем опасны приступы эпилепсии во сне?
- Как помочь пациенту во время приступа?
- Можно ли заниматься спортом?
- Может ли эпилепсия привести к проблемам в учебе, задержке развития, проблем с памятью и поведением?
Введите адрес вашей электронной почты и проверьте почту через 5 минут
***Мы ценим вас и ваше доверие. Наша цель – предоставить вам достоверную информацию о лечении эпилепсии, а также постараться помочь вам или вашим детям жить с этим тяжелым заболеванием. Ни при каких обстоятельствах ваши данные не будут переданы и проданы третьим лицам. Как и вы, мы не любим получать бесполезную почту или рекламу, и постараемся оправдать ваше доверие.
Книга написана простым языком для мам и пап, полна практических советов и рекомендаций эксперта-эпилептолога с мировым именем.
В книге профессора Крамера вы найдёте ответы на многие ваши вопросы об эпилепсии у детей, начиная с видов приступов, правильной диагностики, эффективных методов лечения, и заканчивая практическими советами о том, как повысить качество жизни вашего ребёнка и подготовить его к самостоятельной взрослой жизни.
Читайте также: